Learn Galaxy
Because G-OnRamp is based on the Galaxy platform, the first step to learning how to use G-OnRamp is to acquire some basic familiarity with Galaxy. The “Overview of Galaxy” presentation in the Learning Materials section will give you the necessary basic information about Galaxy. The two screencasts from Galaxy linked here provide an introduction to getting data and comparing genomics features. If you want to learn more about Galaxy, visit the Learn Galaxy page on the Galaxy Wiki.
Workflows
Big picture
We have developed a comprehensive Galaxy workflow that produces multiple complementary datasets to facilitate the annotation of any eukaryotic genome. The entire workflow is shown below.
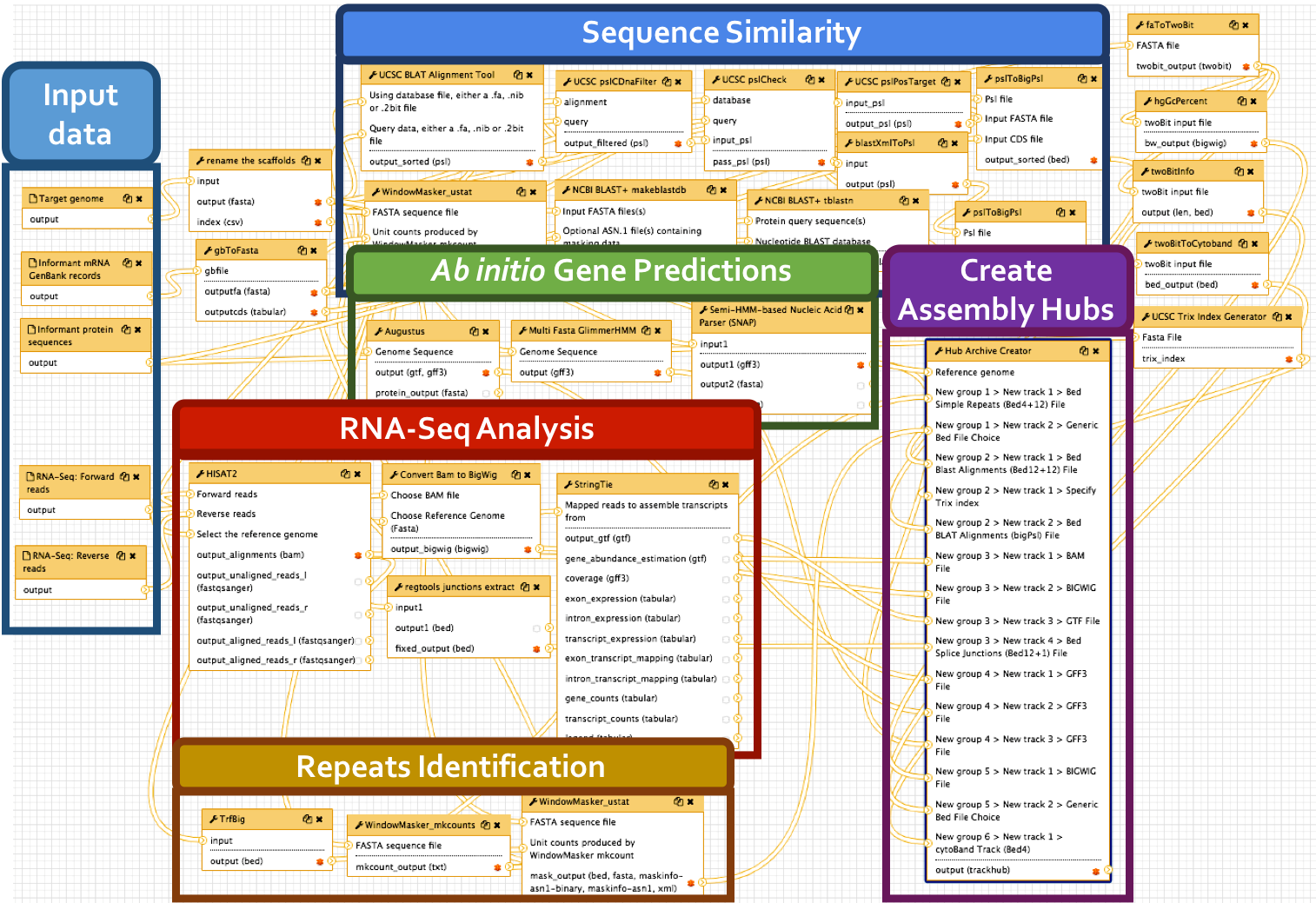
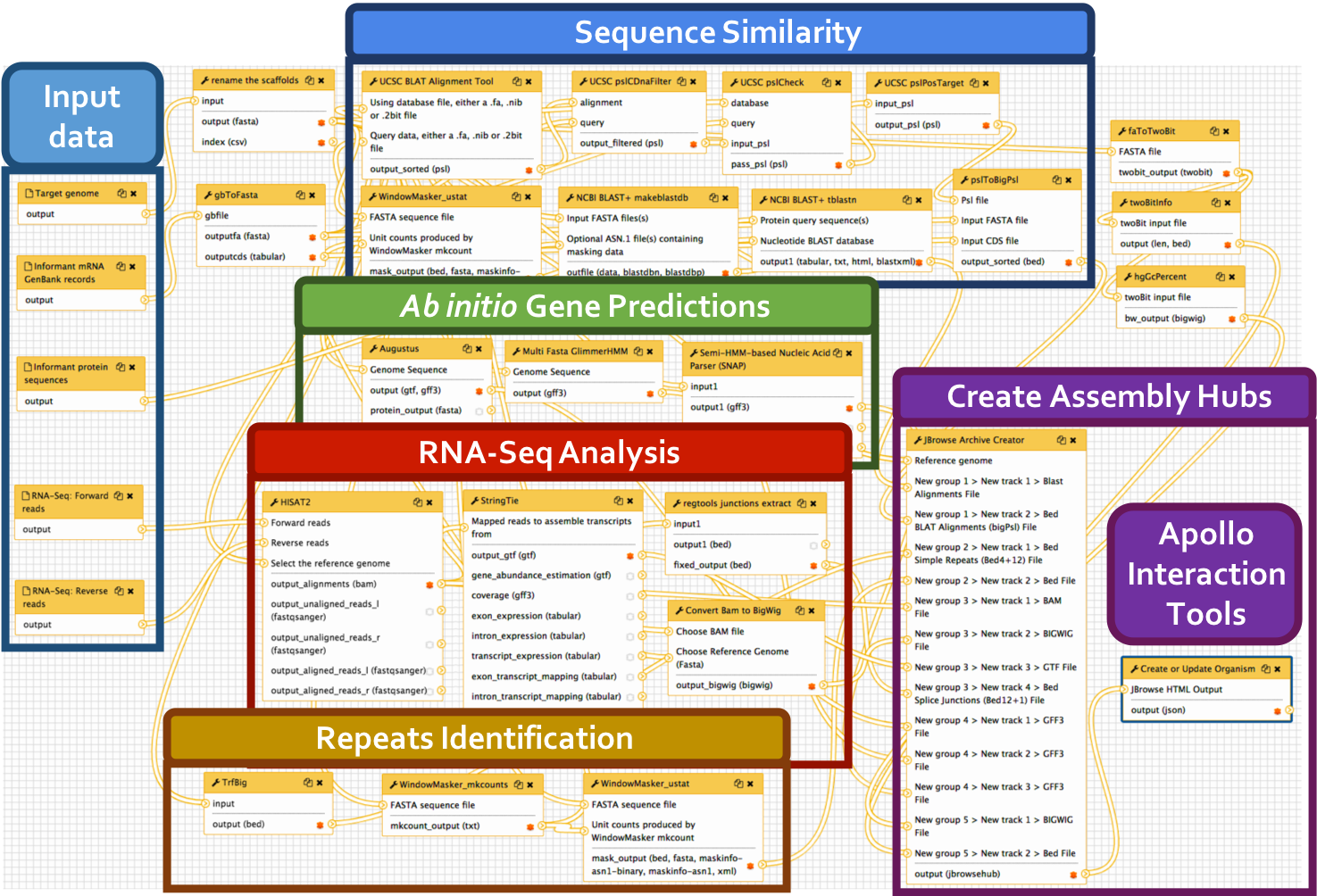
Sub-workflows
The G-OnRamp workflow is divided into four sub-workflows: sequence similarity, repeat regions, RNA-Seq, and gene predictions. These sub-workflows will produce the input datasets for the Hub Archive Creator, which will create the UCSC Genome Browser Assembly Hub.
- BLAST alignment: the genome assembly (in FASTA format) is the input dataset for the NCBI BLAST+ tool makeblastdb, which creates a nucleotide database for BLAST searches. The NCBI BLAST+ tblastn tool searches this nucleotide database against a collection of protein query sequences from an informant species. The blastXmlToPsl and pslToBigPsl tools are used to convert the tblastn search results to the BigPsl format required by the Hub Archive Creator.
- BLAT alignment: RNA GenBank records is the input dataset for the gbToFasta tool, which converts RNA records to FASTA format. The genome assembly (in FASTA format) and RNA records (in FASTA format) are the input datasets for the UCSC BLAT alignment tool, which searches this genome assembly against a collection of RNA query sequences from an informant species. The UCSC pslCDnaFilter tool is used to select near best in genome alignments for each given cDNA and non-comparative, based only on the quality of an individual alignment. The UCSC pslCheck tool is used to validate the PSL output. The UCSC pslPosTarge tool flips psl strands so target is positive and implicit. The pslToBigPsl tool converts the BLAT search results to the BigPsl format required by the Hub Archive Creator.
TrfBig partitions the genome assembly into smaller chunks and then runs Tandem Repeats Finder (TRF) on each chunk to identify tandem repeats within each genomic region. Note that the output of TRF is in BED4+12 format.
RNA-Seq reads are mapped against the genome assembly by HISAT2, and StringTie assembles the mapped RNA-Seq reads into potential transcripts. The “junctions extract” subprogram in Regtools reports the locations of putative introns based on the spliced RNA-Seq reads in the BAM file. The RNA-Seq read coverage track was created by the “Convert BAM to BigWig” tool.
Gene models from three gene predictors (Augustus, GlimmerHMM, and SNAP) were produced using species-specific parameters if they were available. The gene prediction results are converted into the bigGenePred format by the Hub Archive Creator.
Tools we use
Below is a glossary of the tools that we use in the Homology, RNA-Seq, Repeat Regions, and Gene Predictions sub-workflows:
- BLAT mRNA alignments
BLAT
The BLAST-like Alignment Tool (BLAT) is designed to detect sequence similarity between nucleotide and protein sequences. It is originally designed to map transcripts and protein sequences against the genome assembly of the same species. BLAT is much faster but is less sensitive than NCBI BLAST+ (Kent WJ, 2002; PMID: 11932250). BLAT is used in the G-OnRamp workflow to detect sequence similarity between transcript sequences from an informant species and the genome assembly of the target species.
gbToFasta
This UCSC tool converts the transcript sequence records in the GenBank flatfile format into the FASTA format. It also creates an additional file that contains the metadata associated with each transcript, such as the location of the coding region within the transcript.
pslCDnaFilter
This UCSC tool is used to filter the cDNA alignments against a genome assembly produced by BLAT. G-OnRamp uses this tool to filter the transcript alignments produced by BLAT in order to identify the best placement of each transcript from the informant species on the genome assembly of the target species. Transcript alignments with scores that are close to the best score are also kept in order to facilitate the identification of partial genes and paralogs.
pslCheck
This UCSC tool validates the PSL alignment output produced by BLAT. This tool verifies that the PSL file conforms to the PSL specification, and that the sequences and the sequence lengths described in the PSL file are consistent with the lengths of the query and target sequences in the original FASTA files.
pslPosTarget
This UCSC tool converts the PSL alignments so that they are with respect to the orientation of the target sequence.
- NCBI BLAST+
See Camacho C et al., 2009 (PMID: 20003500) for an overview of NCBI BLAST+. See the “BLAST Sequence Analysis Tool” section of the NCBI Handbook for an overview of NCBI BLAST: https://www.ncbi.nlm.nih.gov/books/NBK153387/.
blastXmlToPsl
This UCSC tool converts the XML alignment output file from a tblastn search into the PSL format used by the UCSC Genome Browser.
makeblastdb
Creates a BLAST database from a collection of nucleotide or protein sequences in FASTA format. The BLAST database can include repeat masking data from tools such as WindowMasker and Tandem Repeats Finder (TRF).
pslToBigPsl
This UCSC tool converts the PSL alignment files into the bigPsl format to facilitate the display of sequence alignments in UCSC Assembly Hubs.
tblastn
The tblastn program searches a translated nucleotide database using one or more protein query sequences. For G-OnRamp, tblastn is used to detect sequence similarity between protein sequences from an informant species and the genome assembly of the target species.
Convert BAM to bigWig
This tool calculates the number of reads that aligned to each genomic position, and converts the results into the bigWig format for display on the UCSC Genome Browser and on JBrowse. G-OnRamp uses this tool to show the RNA-Seq alignment coverage.
HISAT2
An alignment program for second and third generation sequencing reads, HISAT2 uses multiple indices to enable the efficient mapping of unspliced and spliced reads against a target genome (Kim D et al., 2015; PMID: 25751142). See the HISAT2 Manual for the list of available alignment options (https://ccb.jhu.edu/software/hisat2/manual.shtml). The G-OnRamp workflow uses HISAT2 to map RNA-Seq reads against the target genome.
regtools junction extract
The junction extract command of regtools (https://github.com/griffithlab/regtools) identifies splice junctions based on the spliced alignments in an RNA-Seq BAM file. The documentation for the regtools junction extract tool is available at https://regtools.readthedocs.io/en/latest/.
StringTie
A reference-based transcriptome assembler, StringTie uses a network flow algorithm to assemble transcripts based on the spliced and unspliced RNA-Seq reads that have been mapped to the target genome (Pertea M et al., 2015; PMID: 25690850). StringTie also provides an estimate of the expression levels of each assembled transcript that could be used to identify differentially expressed genes within a sample.
TrfBig
TrfBig is a wrapper developed by the UCSC Bioinformatics group to facilitate the identification of tandem repeats within genomic sequences using Tandem Repeats Finder (TRF). Tandem repeats are approximate copies of the same DNA sequence that are located next to each other. See Benson G, 1999 (PMID: 9862982) and the Tandem Repeats Finder web site for details (https://tandem.bu.edu/trf/trfdesc.html).
WindowMasker
WindowMasker uses a window-based approach to identify low complexity sequences and transposon remnants within a DNA sequence. The WindowMasker mk_counts module counts the number of times that each (short) sequence of length k (k-mer) that appears in the genome. This information is used by the ustat module to mask repetitive sequences in a genome. The repeat masking information can be incorporated into BLAST database, thereby reducing the number of spurious matches in the BLAST results, and improve the performance of BLAST (Morgulis A et al., 2006; PMID: 16287941).
Augustus
The Augustus gene predictor is based on a Generalized Hidden Markov Model (GHMM), and it can predict multiple isoforms for each gene (Stanke M et al., 2006; PMID: 16845043). Augustus shows high sensitivity and specificity for the gene predictions in many different genomes (http://augustus.gobics.de/accuracy). Augustus also provides a web server for generating species-specific gene prediction parameters (http://bioinf.uni-greifswald.de/webaugustus/trainingtutorial.gsp).
GlimmerHMM
GlimmerHMM is one of the first ab initio gene predictor for eukaryotes and it generally shows higher sensitivity and specificity than Genscan (Majoros WH et al., 2004; PMID: 15145805). The GlimmerHMM user manual is available online at https://ccb.jhu.edu/software/glimmerhmm/man.shtml.
SNAP
SNAP (https://github.com/KorfLab/SNAP) is an ab initio gene predictor for prokaryotic and eukaryotic genomes that shows better sensitivity and specificity than Genscan (Korf I 2004; PMID: 15144565). SNAP can estimate gene prediction parameters for a genome that has limited amount of experimental data or manually curated gene models.
Hub Archive Creator
The Hub Archive Creator converts the genome assembly and the output from repeat finders, sequence similarity searches, RNA-Seq tools, and gene predictors into the UCSC Assembly Hub format (http://genomewiki.ucsc.edu/index.php/Assembly_Hubs). The specification for UCSC Track Hubs and Assembly Hubs are available on the UCSC web site at http://genome.ucsc.edu/goldenPath/help/trackDb/trackDbHub.html. The Hub Archive Creator is available at https://github.com/goeckslab/hub-archive-creator.
JBrowse Archive Creator
The JBrowse Archive Creator converts the genome assembly and the output from repeat finders, sequence similarity searches, RNA-Seq tools, and gene predictors into a format that is compatible with JBrowse. JBrowse (https://jbrowse.org/) is a web-based genome browser where the genome browser image is rendered by the web browser (using HTML5 and JavaScript), which results in improved performance and scalability (Buels R et al., 2016; PMID: 27072794). The JBrowse Archive Creator is available at https://github.com/Yating-L/jbrowse-archive-creator.
Apollo User Manager
Apollo (http://genomearchitect.github.io) is a web-based collaborative genome annotation editor (Lee E et al., 2013; PMID: 24000942). The Apollo User Manager supports multiple operations for managing Apollo user accounts and groups. The currently supported operations include:
- Create a new user
- Delete a user
- Create a user group
- Delete a user group
- Add a user to a group
- Remove a user from a group
Create or Update Organism
This tool takes a JBrowse Archive generated by the JBrowse Archive Creator, and create or update a workspace (organism) on Apollo.
Delete an Apollo record
The tool is used to delete a workspace (organism) from Apollo and export all the gene annotations, genome sequences, and metadata for the organism.
CyVerse interaction via iRODS
This tool transfers the Assembly Hubs produced by G-OnRamp to the CyVerse Data Store (https://www.cyverse.org/data-store). The data transfer is managed by the Integrated Rule-Oriented Data System (iRODS).
faToTwoBit
This UCSC tool converts a genome FASTA sequence file into the twoBit format used by the UCSC Genome Browser (https://genome.ucsc.edu/goldenpath/help/twoBit.html).
hgGcPercent
This UCSC tool uses a sliding window to calculate the GC percentages of a genome assembly (default sliding window size = 5bp with no overlap).
rename the scaffolds
This tool either renames or truncates the scaffold names in the target genome so that they are less than 32 characters. (Older versions of the UCSC Genome Browser do not support sequence identifiers that exceed 31 characters.) The tool takes the target genome in FASTA format as input, and output two files: a renamed target genome file in FASTA format, and a name mapping file which contains the original scaffold names and the renamed scaffold names.
rename the tracks
This tool uses the name mapping file produced by the “rename the scaffolds” tool to rename the scaffolds in a custom track file uploaded by the user. The tool supports custom track files in BED, GFF3, GTF, BAM, and BigWig formats. The renamed custom track file can then be incorporated into the “Hub Archive Creator” and the “JBrowse Archive Creator”.
twoBitInfo
This UCSC tool reports the sequence statistics for a twoBit file (e.g., sequence lengths, gap locations).
UCSC Trix Index Generator
This UCSC tool creates a Trix index to enable searches of free-form text associated with an identifier (https://genome.ucsc.edu/goldenpath/help/trix.html). G-OnRamp uses this tool to create a Trix index of the RefSeq mRNA accession numbers and their sequence descriptions to enable to user to lookup mRNA records by keywords in the UCSC Genome Browser.
Documentation
For detailed G-OnRamp tutorials, see training materials for release 1.1 of G-OnRamp.